
Finding the regulatory sweet spot: the case for clinical input at concept stage
Most digital health teams think of regulation as something that arrives later: after traction, after product-market fit, after the “real” work is done.
In practice, the opposite is true. The earliest decisions you make about intended purpose, feature boundaries, and claims quietly determine whether your product scales smoothly or accumulates hidden regulatory debt that explodes at first release or first update.
What looks like speed early on often turns out to be delay, cost, and loss of leverage later.
This is not about paperwork. It is about classification.
Classification is not a label. It is a design constraint
In the UK and EU, software classification is driven by intended purpose and real-world use, not by what a team meant to build in theory.
At the concept stage, the dominant risk is misalignment: the product behaves one way in the real world, but the team has classified it as something simpler. If this is missed, the cost is usually a forced reclassification months later, when design decisions are already locked in.
At MVP stage, risk increases quietly. Features added for engagement, automation, or “helpfulness” often shift the risk class without anyone explicitly deciding to do so. By the time this is recognised, redesign is expensive and politically painful.
At first release, the risk becomes visible. Claims start to matter. Regulators, partners, and investors begin asking whether the product does what it implies and whether it is allowed to.
By scale and iteration, the danger is now not only initial misclassification but accidental reclassification. Updates trigger new regulatory obligations and teams find themselves doing emergency compliance work just to keep the product live.
These are not edge cases. They are the most common failure pattern we see.
The “sweet spot” problem
There is a persistent myth that lower classification always equals faster market entry. In reality, products that sit too low often struggle to demonstrate value, defensibility, or credibility. They generate weak clinical propositions and limited strategic leverage.
At the other extreme, products that start too high face long timelines, high evidence burdens, and cash constraints before they have learned enough about their users.
Between these sits a narrow but powerful zone: a first release that is viable, defensible, and scalable, without overcommitting the organisation before it needs to.
For many products, this “sweet spot” is Class I or IIa under the UK MDR 2002 and EU MDR, but it is not universal. The correct position depends on the intended final value of the product, not just the first feature set.
Getting this right early creates leverage. Getting it wrong creates drag.
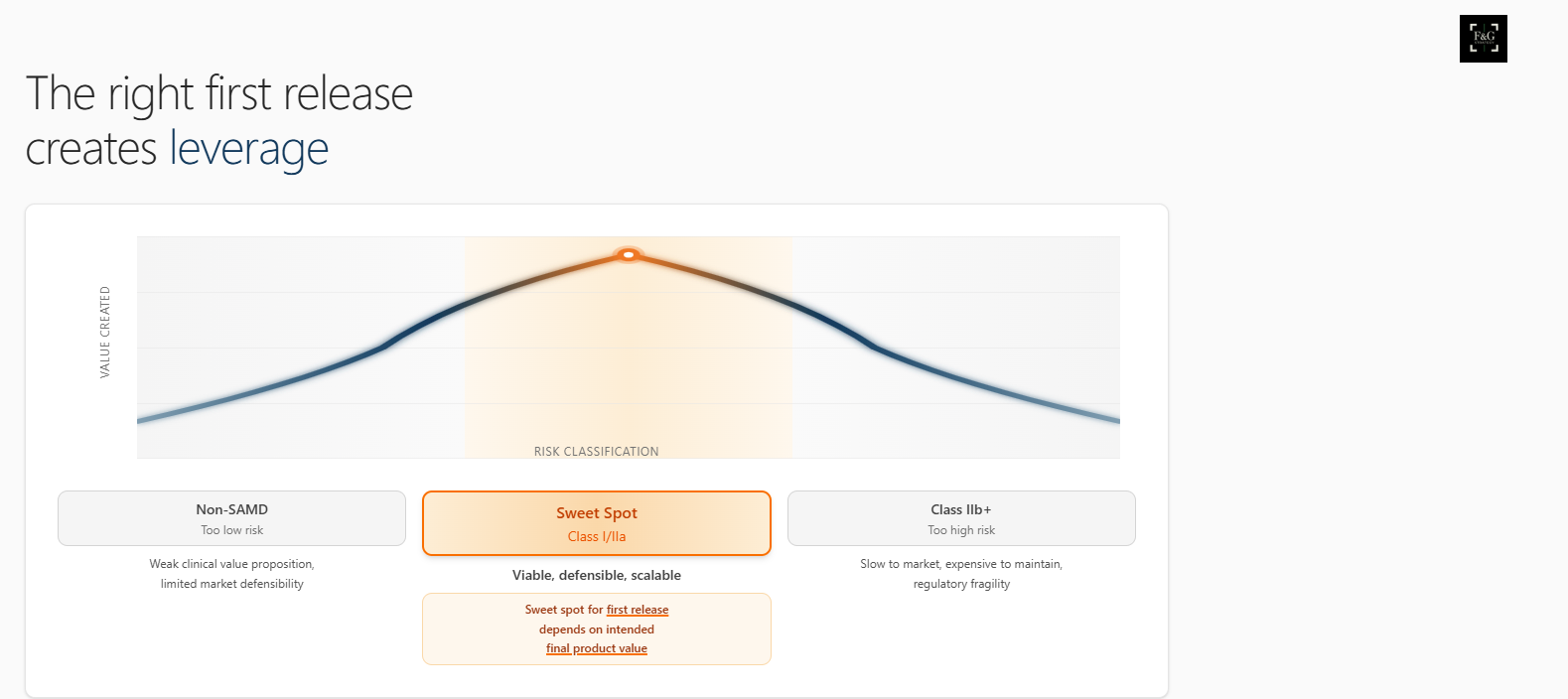
The right first release creates leverage. Too low and you have weak clinical value and limited defensibility. Too high and you face long timelines and high runway risks.
What early alignment actually buys you
When classification, claims, and safeguards are aligned before build and release, several things follow.
Time to market shortens, not because corners are cut, but because the regulatory pathway is predictable. Teams avoid redesign cycles that come from late surprises.
Costs are avoided rather than merely deferred. Rework, emergency consultancy, paused launches, and unplanned evidence generation are expensive forms of learning.
Investor and partner conversations become easier. Fewer uncomfortable questions arise about “what happens if the MHRA disagrees”.
Most importantly, teams retain focus. Energy goes into product evolution, not defensive compliance.
This is judgement under uncertainty, not document production
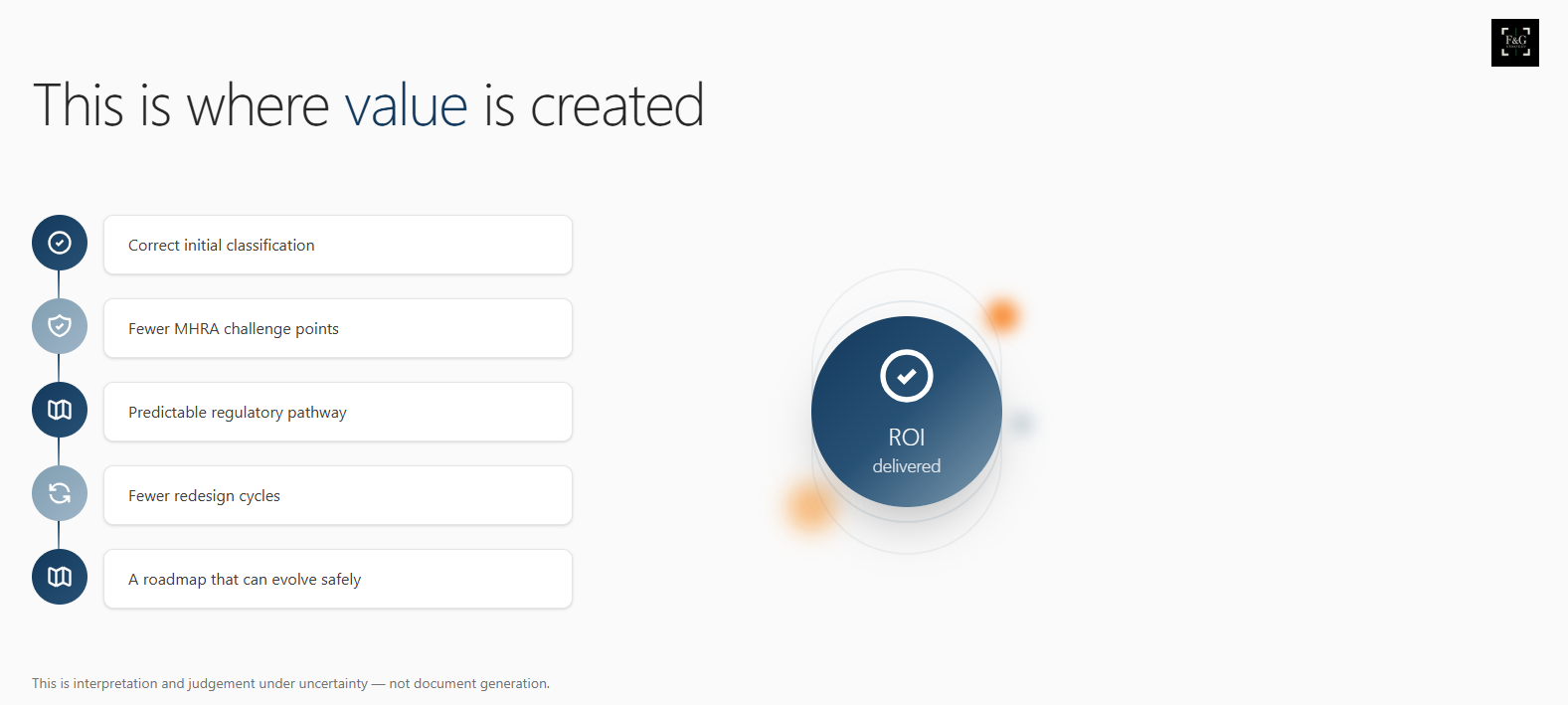
None of this can be reduced to a checklist. Classification and regulatory strategy are interpretive exercises. They involve judgement about behaviour, context, user vulnerability, and how systems are actually used, not just how they are described.
That judgement is hardest at the beginning, when information is incomplete. It is also when it matters most. Clinical expertise in risk formulation makes this judgement more defensible: it provides a structured method for identifying hazards across diagnosis, intervention, monitoring, and referral pathways, and for articulating those risks in terms that regulators, notified bodies, and clinical partners will recognise as credible.
The right first release does not guarantee success. But it does create leverage: financial, regulatory, and strategic.
And leverage, in regulated environments, is the difference between momentum and constant recovery.